Department of Biological Sciences,Graduate School of Science,University of Tokyo
The major ongoing research projects
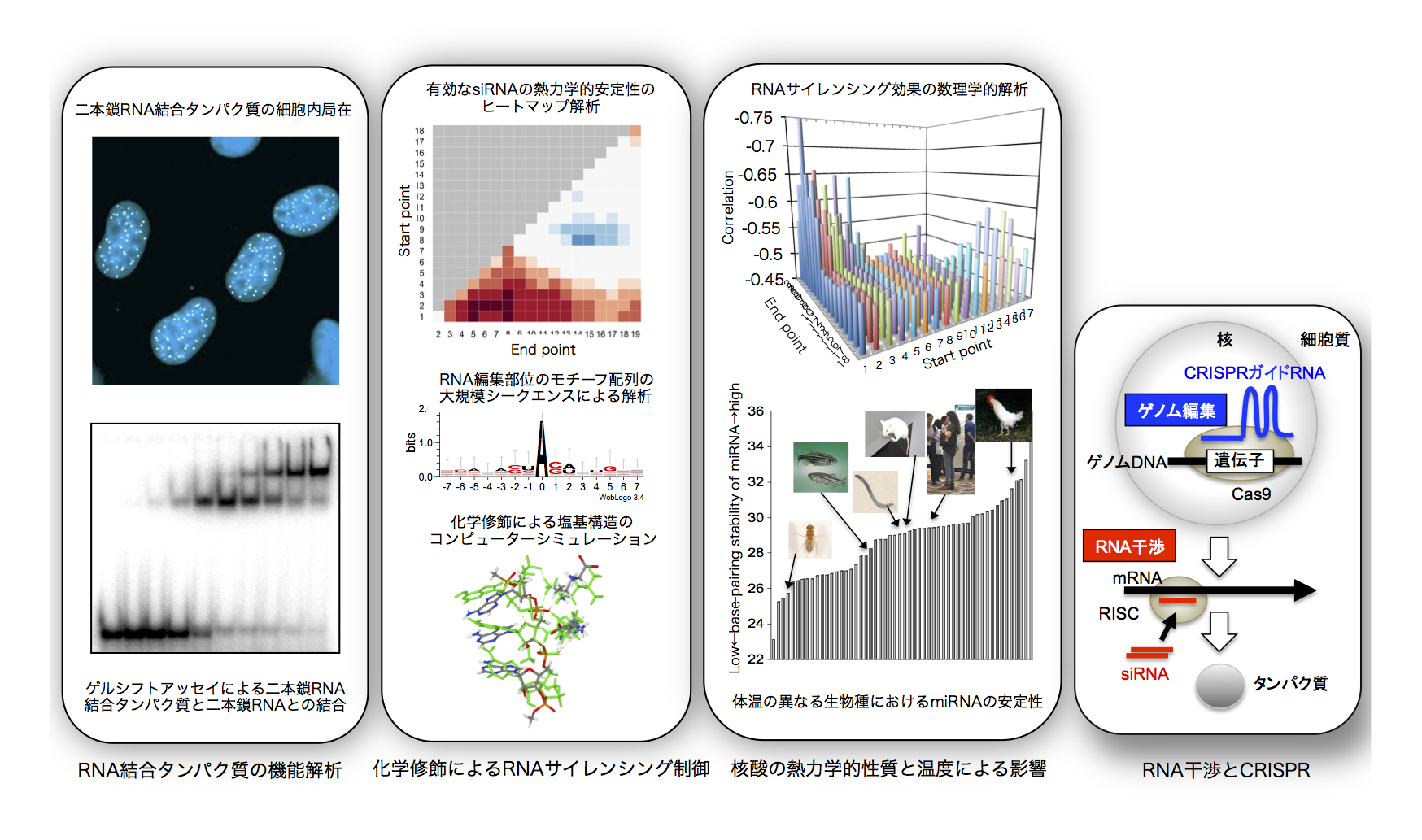
Researches of siRNA&CRISPR
(1) Development of an unique RNA interference approach for disease-related genes and clinical application of siRNA and CRISPR.
(Related papers: Kobayashi et al. 2021, Iribe et al. 2017; Ui-Tei et al. 2008)
(2) The effect of chemical modification on the thermodynamic control of base pairing in RNA interference.
(Related papers: Tian et al. 2019; Ui-Tei. 2016; Hibio et al. 2012; Ui-Tei et al. 2012; Ui-Tei et al. 2008;)
Researches of miRNA
(3) Development of genome-wide application methods and elucidation of molecular mechanisms of genome editing using the CRISPR/Cas9 system.
(Related papers: Ui-Tei et al. 2017 and Naito et al. 2015 )
(4) Elucidation of new human defense mechanisms mediated by microRNAs and their complete gene network through miRNA research.
(Related papers: Takahashi & Ui-Tei 2020; Takahashi et al. 2019; Takahashi et al. 2018; Takahashi et al. 2018; Takahashi et al. 2014; Takahashi et al. 2013)
(5) RNA-binding proteins, like as A-to-I RNA editing enzymes, are used in epitranscriptomic research
(Related papers: Ishiguro et al. 2018; Galipon et al. 2018; Galipon et al. 2017; Kume et al. 2014)
(6) Determining the nuclear function of the miRNA silencing factor TNRC6A and its relationship to cell carcinogenesis
(Related papers: Munakata et al. 2021; Suzawa et al. 2017; Nishi et al. 2015; Nishi et al. 2013)
Main research topics to date
(1) Guidelines for designing siRNA that efficiently knock down a single gene.
(2) Asymmetric region-specific roles of siRNAs.
(3) Construction of siRNA libraries for all human genes
(4) Molecular mechanism of miRNA migration to the cell nucleus
(5) The molecular mechanism of miRNA silencing and its quantification.
Functional analysis of non-coding RNAs is a major challenge for genome science
Genome sequencing projects have revealed that the number of so-called protein-coding "genes" does not differ greatly from lower to higher organisms. If the number of genes in lower organisms does not differ greatly from that in humans, how can complex higher life functions such as thought, emotion, and language be generated? Recent transcriptome analysis has revealed that higher eukaryotes, including humans, contain a large number of RNAs called non-coding RNAs (non-protein-coding RNAs). The strong correlation between the number and diversity of non-coding RNAs and the complexity of living organisms suggests that these RNAs may be deeply involved in the higher functions of living organisms, and clarifying the functions of non-coding RNAs has become a major issue in genome science.
Various environmental factors regulate genetic information via RNA.
Our bodies are composed of cells, which are the most basic building blocks of life. In the cell, RNA is transcribed from DNA, which constitutes the genome, and proteins are translated from RNA, which is called the central dogma. However, as the genome project has progressed and the overall picture of genome information has become clearer, it has become clear that life functions are not determined solely by the genome, but that factors derived from the environment are also important. In other words, complex and higher-order life phenomena and age-related changes in gene expression are greatly influenced by environmental factors such as light, gravity, radiation, temperature, microbial infection, and stress. In recent years, research on RNA has progressed rapidly, and it has become clear that RNA is not merely an intermediary for genetic information, but also exhibits a variety of sequence- and structure-dependent functions by taking advantage of its flexible nature. Such non-coding RNAs are considered to be different from the RNAs that have been the subject of research so far. RNA is also attracting attention as a molecule that constructs a new process of gene expression regulation called epitranscriptome by various chemical modifications. In this laboratory, we are investigating new mechanisms of gene regulation that successfully convey the effects of the environment, focusing on RNA.
(1) Guidelines for designing siRNAs that can efficiently knock down one gene specifically
RNAi is a phenomenon in which double-stranded RNA (dsRNA), which is homologous to a specific region of a gene, cleaves the homologous part of mRNA. However, in most mammalian cells, when dsRNAs longer than about 30 base pairs enter the cell, the interferon response, a biological defense mechanism against viruses, is induced, leading to cell death. However, we have shown that RNAi with long dsRNA can be induced in mammals in limited cell systems such as embryonic stem cells, and that RNAi machinery exists in mammalian cells as well.
The long dsRNA was cleaved by RNase III enzyme, Dicer, into short siRNA with two bases protruding from the 3' end. siRNA, the cleavage product, was shown to be capable of inducing RNAi in mammalian cells without causing an interferon response. The RNAi method has been attracting attention as a useful gene function analysis method suitable for the post-genome sequencing era because it is simple to operate and requires only a minimum of 19 nucleotides to be sequenced. However, we have found that in mammalian cells, siRNAs of any sequence can not induce RNAi, and whether RNAi occurs or not depends largely on the siRNA sequence. Based on RNAi experiments using cultured cells against reporter genes and endogenous genes, as well as various experiments such as RNAi in individuals and DNA-vectored RNAi, it was considered that the sequences of siRNAs that are effective satisfy all of the following four conditions.
(1) The 5' end of the guide strand is A or U. (2) The 5' end of the passenger strand is G or C. (3) The 5' region of the antisense strand contains a large number of A or U. (4) There is no long GC sequence. We have constructed a website (siDirect) for designing siRNA sequences that meet these requirements for effective sequences and that have at least two base mismatches with all genes other than the target. This is now available on the web for anyone to use.
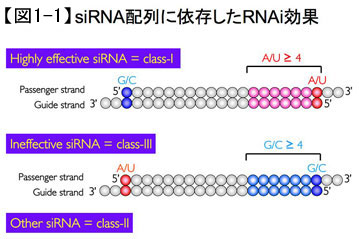
Why are siRNAs that meet the above requirements more effective in RNAi? There was a striking asymmetry between the siRNAs with high and low RNAi efficacy, as shown in Fig. 1. siRNAs unravel from their double-stranded form to become single-stranded during RNAi execution, and are incorporated into the RNA-induced silencing complex (RISC) to pair with the target mRNA.
A- or U-pairs of double-stranded RNA are more unstable than G- or C-pairs, and A- and U-pairs are more prone to unraveling. Therefore, the abundance of A or U in the 5' region of the guide strand of siRNAs that work suggested that siRNAs would asymmetrically unwind from the 5' side of the guide strand to the single strand. On the other hand, we are the first to identify the human and mouse genes for Argonaute (Ago), the core protein of RISC, and recent structural studies of thermophilic bacterial Ago indicate that the six bases in the 5' end region of the siRNA guide strand are Since the end of the guide strand that is anchored to the Ago is considered to be the end of the first single strand, it is possible that siRNAs that satisfy the aforementioned conditions can efficiently induce RNAi because the guide strand is a sequence that can be efficiently incorporated into the RISC complex. Therefore, siRNAs that meet the aforementioned conditions can efficiently induce RNAi because the guide strand is a sequence that can be efficiently incorporated into the RISC complex.
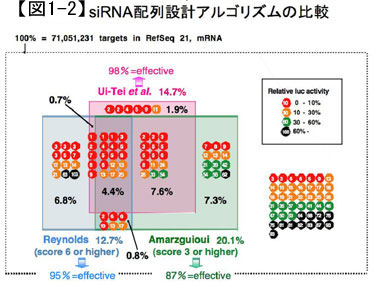
Although several other effective siRNA sequence design algorithms have been proposed, we examined the efficacy of three widely used algorithms, including ours, using a reporter assay targeting the firefly luciferase gene. We examined the efficacy of three widely used algorithms worldwide, including our method, using a reporter assay targeting the firefly luciferase gene. The results showed that 98% of the siRNAs selected by our algorithm gave the best results, suppressing the amount of mRNA of the target gene to 33% or less (Figure 1-2). Since our method can select siRNA sequences that are effective for 99.5% of human genes, we believe that we have almost established the basic technology for RNAi in mammals, including humans, through this study.
However, in RNAi, it has been found that genes other than the target gene are sometimes suppressed even though siRNA with a 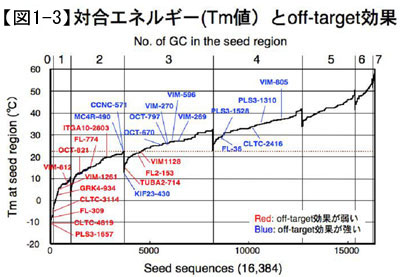 sequence that is completely complementary to the target gene is used. Using microarray data, we analyzed the mechanism by which the off-target effect occurs, and obtained results suggesting that genes whose RNA sequences are complementary only to the seventh base of the siRNA guide strand from the 5' end to the second to eighth base (called the seed region) are suppressed by the off-target effect. In other words, in the case of siRNA, the full-length gene is suppressed by the off-target effect. In other words, in the case of siRNA, genes with completely complementary sequences throughout their entire length (target genes) are suppressed by the RNAi effect, but not only that, genes with complementary sequences only in the seed region (non-target genes) may also be suppressed by an unintended effect called the off-target effect. In order to knock down only one gene of interest by RNAi, a method that does not cause the off-target effect is necessary, but a random 7-base sequence has a probability of appearing once in 47 = 16,384 bases. However, since the off-target effect occurs differently depending on the siRNA sequence, we thought that factors other than the sequence are involved. In other words, since the off-target effect is caused by base pairing between the seed region of siRNA and the target mRNA, we thought that the pairing energy of this base pairing would be a major factor controlling the degree of the off-target effect, and conducted research to demonstrate this. Using two measures of the pairing energy, melting temperature (Tm) and free energy (Gibbs free energy change, ΔG), we found that the Tm value of double-stranded RNA, even with only seven bases, was significantly higher than that of double-stranded RNA. As expected, the off-target effect was stronger for RNAs with higher Tm values in the seed region, while the off-target effect was hardly observed for RNAs with lower Tm values (Fig. 1-3).
sequence that is completely complementary to the target gene is used. Using microarray data, we analyzed the mechanism by which the off-target effect occurs, and obtained results suggesting that genes whose RNA sequences are complementary only to the seventh base of the siRNA guide strand from the 5' end to the second to eighth base (called the seed region) are suppressed by the off-target effect. In other words, in the case of siRNA, the full-length gene is suppressed by the off-target effect. In other words, in the case of siRNA, genes with completely complementary sequences throughout their entire length (target genes) are suppressed by the RNAi effect, but not only that, genes with complementary sequences only in the seed region (non-target genes) may also be suppressed by an unintended effect called the off-target effect. In order to knock down only one gene of interest by RNAi, a method that does not cause the off-target effect is necessary, but a random 7-base sequence has a probability of appearing once in 47 = 16,384 bases. However, since the off-target effect occurs differently depending on the siRNA sequence, we thought that factors other than the sequence are involved. In other words, since the off-target effect is caused by base pairing between the seed region of siRNA and the target mRNA, we thought that the pairing energy of this base pairing would be a major factor controlling the degree of the off-target effect, and conducted research to demonstrate this. Using two measures of the pairing energy, melting temperature (Tm) and free energy (Gibbs free energy change, ΔG), we found that the Tm value of double-stranded RNA, even with only seven bases, was significantly higher than that of double-stranded RNA. As expected, the off-target effect was stronger for RNAs with higher Tm values in the seed region, while the off-target effect was hardly observed for RNAs with lower Tm values (Fig. 1-3).
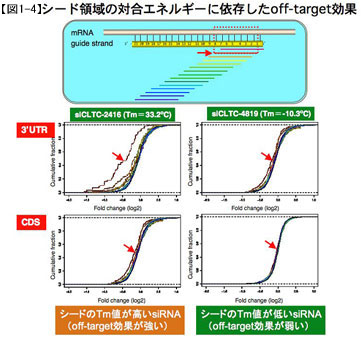
The same trend was observed for ΔG, but a stronger correlation was observed when Tm was used. In addition, microarray results showed that genes whose complementary sequence to the seed was in the 3'UTR were strongly repressed, while genes whose complementary sequence was in the CDS were repressed to a lesser extent (Fig. 1 - 4). 4).
However, in both cases, there was a similar strong correlation between the Tm value of the seed and the repression effect, suggesting that the strength of the off-target effect of siRNA depends on the congruent energy. Therefore, the reason for the stronger inhibitory effect of the target site in the 3'UTR is thought to be on the mRNA side, not on the siRNA itself.
From these studies, the main contribution of the siRNA sequence to the mechanism of action of small RNAs has been clarified (Fig. 1-5). The characteristics of siRNA sequences with high RNAi effect were thought to be mainly related to the step of incorporation of the guide strand into RISC. Furthermore, it became clear that the off-target effect could be avoided by thermodynamic destabilization of the pairing of the seed region with the target gene. We have also constructed a web server that enables us to design siRNA sequences with such Tm in mind, and have released it as siDirect 2.0.
2) Asymmetric region-specific roles of siRNAs
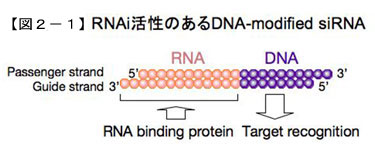
As one of the approaches to elucidate the mechanism of action of small RNAs, we investigated the differences in the roles of different regions of siRNAs, which are very short (about 21 bases). In a series of experiments in which a portion of the siRNA was systematically replaced with DNA, we found that replacing all of the 5'-end region of the siRNA guide strand (8 bases from the 5'-end) had no effect on RNAi activity, but replacing the remaining 2 bases on the 3 The remaining 2/3 of the 5' end of the siRNA was replaced with DNA, even if only a few bases were replaced. The remaining 2/3 of the 5' end of the siRNA is almost identical to the "seed" region (2-8 bases from the 5' end) described above. Our results showed that the RNAi effect of siRNA with the seed region replaced by DNA was almost unchanged, suggesting that the role of the seed region of siRNA for the gene that will eventually be paired with the full-length gene is to first identify and pair with the target sequence, and for this purpose, 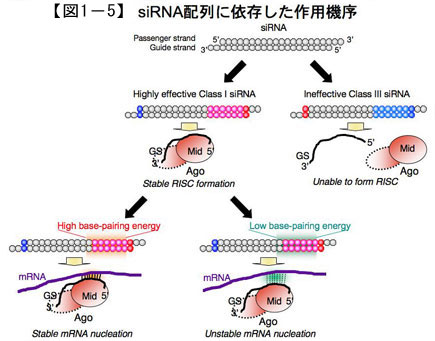 the seed region does not necessarily have to be RNA, but rather DNA. Therefore, the seed region does not necessarily have to be RNA, but can be DNA as well (Fig. 2-1).
the seed region does not necessarily have to be RNA, but rather DNA. Therefore, the seed region does not necessarily have to be RNA, but can be DNA as well (Fig. 2-1).
However, for non-target genes for which only the seed has a complementary sequence, only the seed portion is considered to act as the point of action. Since RNA-RNA pairing is stronger than RNA-DNA pairing, a strong off-target effect on non-target genes is expected to occur in the case of siRNA, whose seed is RNA, due to RNA-RNA pairing with target genes. However, by replacing this part with DNA, it is thought to be weakened.Since RNA-RNA pairing is stronger than RNA-DNA pairing, the repressive effect on non-target genes is expected to be weakened by replacing this part with DNA. On the other hand, the 3' end of the guide strand almost completely loses its RNAi activity when it is replaced by DNA. Based on binding experiments with RLC (RISC-loading complex) or RNA-binding proteins contained in RISC, this region was considered to function as a binding site for double-stranded RNA-binding proteins. The role of this area is under investigation.